hemofilia
i Martina Feichter, redaktor medyczny i biologDr. med. Fabian Sinowatz jest freelancerem w redakcji medycznej
Więcej o ekspertachMartina Feichter studiowała biologię w aptece przedmiotowej w Innsbrucku, a także zanurzyła się w świecie roślin leczniczych. Stamtąd nie było daleko do innych tematów medycznych, które do dziś urzekają ją. Szkoliła się jako dziennikarka w Akademii Axel Springer w Hamburgu, a od 2007 roku pracuje dla - najpierw jako redaktor, a od 2012 jako niezależny pisarz.
Więcej o ekspertach Wszystkie treści są sprawdzane przez dziennikarzy medycznych.
Hemofilia (choroba krwi) to zaburzenie krzepnięcia krwi, które jest zwykle dziedziczne. Osoby cierpiące na tę chorobę nie mają ważnych czynników krzepnięcia krwi lub mają wadę. W rezultacie hemofile mają skłonność do krwawień i siniaków. Hemofilia nie została jeszcze wyleczona. Jednak dzięki nowoczesnym terapiom chorzy na hemofilię mogą prowadzić w dużej mierze normalne życie. Przeczytaj wszystko, co musisz wiedzieć o hemofilii tutaj.
Kody ICD dla tej choroby: Kody ICD to uznane na całym świecie kody diagnoz medycznych. Można je znaleźć np. w pismach lekarskich czy na zaświadczeniach o niezdolności do pracy. D66D67D68
Krótki przegląd
- Co to jest hemofilia? Genetyczne zaburzenie krzepnięcia krwi (koagulopatia). Nazywa się to również hemofilią.
- Formy hemofilii: Najczęstszą jest hemofilia A, a następnie hemofilia B. Inne postacie, takie jak hemofilia C, zespół von Willebranda-Jürgensa i parahemofilia są mniej powszechne.
- Przyczyna: Niedobór lub defekt czynników krzepnięcia (białka krwi niezbędne do krzepnięcia krwi). Jest w większości dziedziczna i rzadko nabyta (spowodowana spontaniczną mutacją genu).
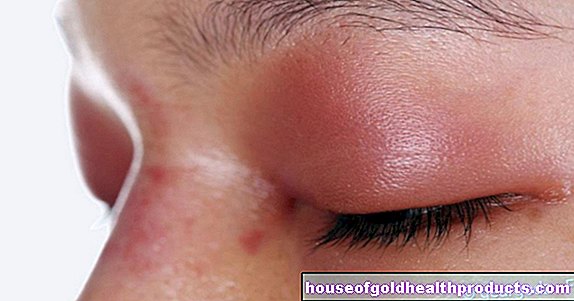
- Objawy: zwiększona skłonność do krwawień, które łatwo mogą prowadzić do krwawień i siniaków (krwiaki). Krwawienie trwa również dłużej niż zwykle. Stopień nasilenia objawów zależy od ciężkości hemofilii.
- Diagnostyka: Pomiar różnych parametrów krwi (aPTT, Quick value, czas trombinowy osocza, czas krwawienia, liczba płytek krwi), określenie aktywności czynników krzepnięcia
- Leczenie: uzupełnienie brakującego czynnika krzepnięcia (w postaci koncentratów czynnika); w niektórych przypadkach inne leki (takie jak desmopresyna na łagodną hemofilię A)
Hemofilia: opis
Hemofilia to wrodzone zaburzenie krzepnięcia krwi. Osoby dotknięte chorobą (hemofiliacy, „hemofiliacy”) nie mogą tworzyć wystarczająco funkcjonalnych czynników krzepnięcia. Są to białka we krwi, które są niezbędne do krzepnięcia krwi. Ze względu na brak czynników krzepnięcia, skrzepy krwi i rany nie mogą się łatwo tworzyć. Dlatego pacjenci z hemofilią są podatni na krwawienie. Mają też rany, które krwawią dłużej niż normalnie. W pewnych okolicznościach może to być niebezpieczne.
Termin medyczny określający zaburzenie krzepnięcia krwi ze zwiększoną tendencją do krwawień to „skaza krwotoczna”. Ogólna nazwa zaburzenia krzepnięcia to „koagulopatia”.
Hemofilia: częstotliwość
Hemofilia występuje prawie wyłącznie u chłopców i mężczyzn. Jest to stosunkowo rzadkie: tylko około dwóch na 10 000 mężczyzn ma hemofilię. W Niemczech dotkniętych jest około 10 000 osób. Około 3000 do 5000 z nich ma ciężką postać choroby.
Hemofilia: formy
Istnieją różne czynniki krzepnięcia. W zależności od tego, na który czynnik krzepnięcia wpływa hemofilia, lekarze rozróżniają różne postacie choroby.
Hemofilia A.
W hemofilii A występują problemy z czynnikiem krzepnięcia VIII (globulina antyhemofilna A): Albo organizm nie jest w stanie wyprodukować go w wystarczających ilościach, albo jest uszkodzony. Około 85 procent wszystkich chorych na hemofilię cierpi na hemofilię A. Prawie wszyscy z nich to mężczyźni.
Hemofilia B.
W hemofilii B brakuje czynnika krzepnięcia IX (globulina antyhemofilna B lub czynnik bożonarodzeniowy). Tutaj również pacjenci to głównie mężczyźni. W przeszłości hemofilia B występowała częściej w angielskiej rodzinie królewskiej i rosyjskiej rodzinie carskiej. Dlatego nazywana jest również „chorobą królów”.
Inne formy hemofilii
Oprócz głównych postaci hemofilii A i B istnieją inne dziedziczne zaburzenia krzepnięcia. Jednym z nich jest zespół von Willebranda-Jürgensa, zwany także zespołem von Willebranda (vWS). Skupiamy się tutaj na czynniku von Willebranda: ten czynnik krzepnięcia jest albo za mały, albo wadliwy. Podobnie jak w przypadku hemofilii A i B, prowadzi to do zwiększonej tendencji do krwawień (skaza krwotoczna). W Niemczech zespół von Willebranda-Jürgensa występuje nawet u jednego procenta populacji. W przeciwieństwie do dwóch głównych postaci hemofilii, to zaburzenie krzepnięcia w równym stopniu dotyka mężczyzn i kobiety.
W rzadkich przypadkach występują również inne zaburzenia krzepnięcia, które wynikają z braku czynników krzepnięcia. Mają też zwiększoną skłonność do krwawień (skaza krwotoczna). Obejmują one:
- Hemofilia C: Niedobór czynnika czynnościowego XI
- Parahemofilia: brak funkcjonującego czynnika V
- Hipoprokonwertynemia: Niedobór funkcjonalnego czynnika VII
- Niedobór czynnika roboczego Stuarta Prowera: Brak czynnika roboczego X
- Zespół Hagemana: niedobór funkcjonalnego czynnika XII
- Niedobór fibrynazy: niedobór funkcjonalnego czynnika XIII
Hemofilia: objawy
Objawy hemofilii A (niedobór czynnika VIII) i hemofilii B (niedobór czynnika IX) są takie same – nawet jeśli w każdym przypadku dotyczy to innych czynników krzepnięcia. Ogólnie można powiedzieć: im mniej funkcjonujących czynników krzepnięcia, tym wyraźniejszy obraz kliniczny.
Hemofilia: stopnie nasilenia
Nasilenie hemofilii zależy od tego, jak bardzo aktywność czynników krzepnięcia jest zmniejszona w porównaniu z ich funkcją u zdrowych ludzi. Jeśli aktywność czynnika jest tylko nieznacznie zmniejszona, osoby dotknięte chorobą zwykle nie mają żadnych objawów. Z drugiej strony, u objawowych hemofilów aktywność czynnika jest bardziej zmniejszona, co powoduje bardziej wyraźne objawy. Istnieją trzy stopnie nasilenia hemofilii (A i B):
Łagodna hemofilia:Aktywność czynnika wynosi od 6 do 45 procent normalnej aktywności zdrowej osoby; około 20 procent wszystkich osób z hemofilią ma ten stopień nasilenia. Osoby dotknięte chorobą często nie zauważają wiele ze swojej choroby. Właśnie dlatego hemofilię wykrywa się u wielu osób w wieku młodzieńczym lub dorosłym, gdy operacje lub poważne urazy krwawią dłużej niż oczekiwano.
Umiarkowana hemofilia: Aktywność czynnika wynosi od 1 do 5 procent normalnej aktywności; około 20 procent wszystkich hemofilów jest również dotkniętych. Objawy zwykle ujawniają się w ciągu pierwszych kilku lat życia z niezwykle długim krwawieniem i częstymi siniakami. Podobnie jak w przypadku łagodnej hemofilii, krwawienie jest zwykle wynikiem urazu lub zabiegu chirurgicznego. Spontaniczne krwawienie jest rzadkie.
Ciężka hemofilia: Aktywność czynnika wynosi mniej niż 1 procent normalnej aktywności. Dotyczy to około 55 procent wszystkich chorych na hemofilię. Hemofilia jest zwykle zauważalna już po urodzeniu: usunięcie pępowiny wywołuje masywne krwawienie. Ciężkie krwawienia z nosa nie są rzadkością w okresie niemowlęcym. Ponadto nawet najmniejsze urazy czy stłuczki mogą powodować obfite krwawienia pod skórą (siniaki). Krwawienie wewnętrzne jest również częstsze, na przykład bolesne krwawienie do dużych stawów (takich jak kolana, łokcie). Zazwyczaj wiele krwawień nie ma wyraźnej przyczyny (krwawienie samoistne).
Hemofilia: ryzyko i niebezpieczeństwa
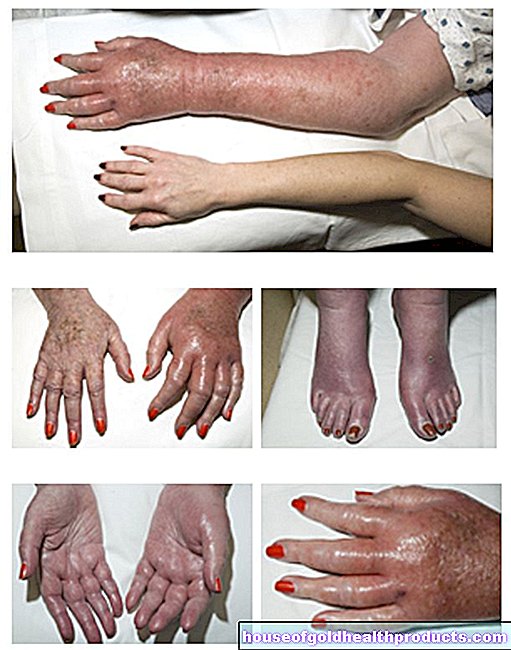
Krwawienie do stawów (hemarthrosis) często występuje wielokrotnie, zwłaszcza w ciężkiej hemofilii. W rezultacie dotknięty staw może się deformować, przedwcześnie zużywać (choroba zwyrodnieniowa stawów) i stopniowo usztywniać. Osoby z zaawansowaną hemartozą ledwo poruszają rękami i nogami i są uzależnione od wózka inwalidzkiego.
Innym niebezpieczeństwem związanym z hemofilią jest krwawienie do mięśni: może uszkodzić tkankę mięśniową i prowadzić do osłabienia mięśni.
Zasadniczo krwawienie wewnętrzne w hemofilii może wystąpić w dowolnym obszarze narządów. Krwawienie w mózgu jest rzadkie, ale niebezpieczne: na przykład może upośledzać zdolność myślenia i osłabiać koncentrację. Ciężki krwotok mózgowy może być nawet śmiertelny! Krwawienie w jamie brzusznej może w pewnych okolicznościach zagrażać życiu. Silne krwawienie w części ustnej gardła może wpływać na drogi oddechowe.
Oprócz skłonności do krwawień (skaza krwotoczna), hemofilia o dowolnym nasileniu może również prowadzić do zaburzeń gojenia ran.
Zespół von Willebranda-Juergensa: objawy
Osoby z zespołem von Willebranda-Jürgensa mają również zwiększoną skłonność do krwawień. Najczęściej występuje lekkie krwawienie pod skórą (siniaki), krwawiące dziąsła lub przedłużone krwawienie po zabiegu chirurgicznym, np. po ekstrakcji zęba. Ale są też pacjenci, którzy cierpią na obfite krwawienie.
Hemofilia: leczenie
Terapia hemofilii zależy od rodzaju i ciężkości hemofilii. Lekarz prowadzący opracuje dla każdego pacjenta odpowiedni plan terapii. Oprócz leczenia farmakologicznego może również zalecić ogólne środki. Na przykład wskazane może być, szczególnie w przypadku ciężkiej hemofilii, zachowanie fizycznej opieki i unikanie niektórych (szkodliwych) sportów.
Hemofilia A i B
Obecnie do leczenia hemofilii dostępne są tak zwane koncentraty czynników. Są to koncentraty czynników krzepnięcia krwi VIII (dla hemofilii A) lub IX (dla hemofilii B) uzyskane z osocza krwi lub poddane inżynierii genetycznej. Muszą być wstrzykiwane do żyły (dożylnie). Wielu cierpiących uczy się samodzielnie wstrzykiwać ten czynnik. Daje im to dużą niezależność w radzeniu sobie z chorobą.
W latach 60. i 70. wiele osób w Niemczech zaraziło się zapaleniem wątroby i/lub wirusem HIV z powodu preparatów skażonych czynników. Dziś to już praktycznie nie może się zdarzyć: osocze krwi jest ściśle kontrolowane i poddawane wstępnej obróbce przed podaniem. A dzięki koncentratom czynników genetycznie modyfikowanych generalnie nie ma ryzyka infekcji.
W przypadku łagodnej i umiarkowanej hemofilii podanie koncentratu czynnika jest konieczne tylko w razie konieczności (leczenie na żądanie): Antykoagulant podaje się np. w przypadku większego krwawienia lub przed planowaną operacją. Drobne urazy, takie jak otarcia, nie wymagają leczenia koncentratem czynnika. Krwawienie można zwykle zatrzymać, stosując lekki nacisk na krwawiące miejsce.
Z drugiej strony osoby z ciężką hemofilią muszą regularnie wstrzykiwać sobie koncentrat czynnika (leczenie długoterminowe). Czynnik VIII w hemofilii A podaje się dwa do trzech razy w tygodniu. Czynnik IX w hemofilii B zazwyczaj musi być wstrzykiwany tylko raz lub dwa razy w tygodniu ze względu na jego dłuższy czas retencji we krwi.
Operacje i ostre urazy
Przed operacją (nawet jeśli planowana jest ekstrakcja zęba) u wszystkich chorych na hemofilię należy przeprowadzić terapię przygotowawczą. W tym celu zwykle podaje się koncentrat czynnika. To jedyny sposób, aby zapobiec poważnym powikłaniom spowodowanym nadmierną utratą krwi podczas operacji.
W przypadku łagodnej hemofilii A zamiast koncentratów czynnika można przed planowaną operacją podać lek stabilizujący krzepliwość krwi. Obejmuje to na przykład desmopresynę. To jest sztucznie wyprodukowane białko. Stymuluje uwalnianie zmagazynowanego czynnika VIII z naczyń krwionośnych. Lek można stosować tylko przez kilka dni, w przeciwnym razie sklepy wkrótce będą puste.
Wskazówka: przed planowanym zabiegiem osoby z hemofilią powinny przedyskutować ze swoim hematologiem lub ośrodkiem hemofilii, która terapia profilaktyczna jest wskazana w ich przypadku.
W przypadku ostrych urazów w nagłych wypadkach, oprócz miejscowych zabiegów hemostazy (np. bandaże uciskowe) konieczne jest również podanie koncentratu czynnika.
Koncentrat czynnika: powikłania
Niektórzy ludzie tworzą przeciwciała (inhibitory) przeciwko czynnikom krzepnięcia w koncentracie czynnika. Ta tak zwana hemofilia inhibitorowa występuje znacznie częściej u osób z hemofilią A niż u osób z hemofilią B. Inhibitory dezaktywują dodany czynnik krzepnięcia. Terapia nie jest wtedy tak skuteczna, jak jest to pożądane. Hemofilia B zagraża również ciężkim reakcjom alergicznym i innym powikłaniom.
Ilość inhibitorów we krwi podawana jest w tak zwanej jednostce Bethesda (BE). Im wyższa wartość BE, tym więcej inhibitorów znajduje się we krwi pacjenta.
W hemofilii A niewielkie nagromadzenie inhibitorów można często skompensować przez zwiększenie dawki koncentratu czynnika. Jeśli inhibitory powstają w dużych ilościach, zaleca się terapię tolerancji immunologicznej: pacjent otrzymuje bardzo wysokie dawki brakującego czynnika krzepnięcia w złożonym schemacie leczenia. Układ odpornościowy powinien stopniowo przyzwyczajać się do jego obecności i powstrzymywać powstawanie inhibitorów.
Rzadki inhibitor hemofilii w hemofilii B jest traktowany inaczej. Na przykład osoby dotknięte chorobą otrzymują leki wpływające na układ odpornościowy (immunomodulatory).
Lek przeciwbólowy
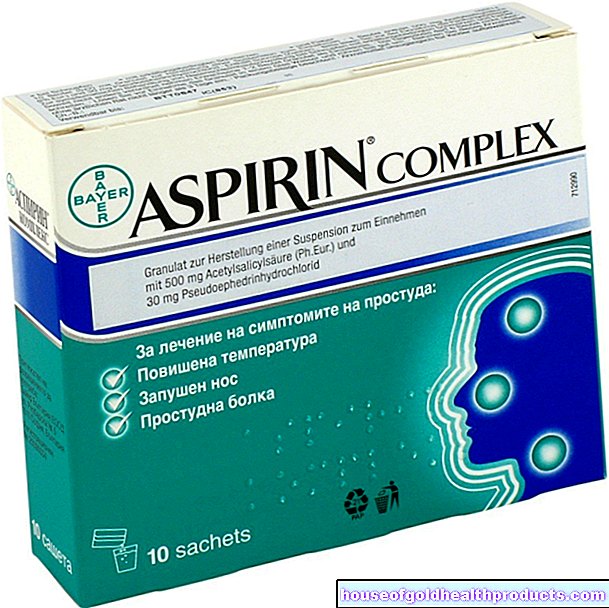
Poważna hemofilia może powodować silny ból u osób dotkniętych chorobą. Na przykład krwawienie do stawów może być bardzo bolesne. Wtedy pomagają leki przeciwbólowe, takie jak ibuprofen. Przeciwnie, łagodzący ból kwas acetylosalicylowy (ASA) nie nadaje się do hemofilii: jeszcze bardziej zwiększa skłonność do krwawień (efekt uboczny ASA).
Zespół von Willebranda-Juergensa (VWS)
W zespole von Willebranda-Jürgensa rozróżnia się różne typy, które są leczone w różny sposób: W tak zwanym typie 1 substancję czynną desmopresynę podaje się w razie potrzeby (przed operacją lub w przypadku ostrego krwawienia). Stymuluje uwalnianie nagromadzonych czynników krzepnięcia.
Desmopresynę stosuje się również w typie 2; jednak lek nie zawsze działa tutaj. Następnie lekarz przepisuje zamiast tego koncentrat czynnika (z czynnikiem von Willebranda).
Pacjenci z VWS typu 3 są zawsze leczeni koncentratem czynnika.
Hemofilia: przyczyny i czynniki ryzyka
Hemofilia to wrodzona choroba genetyczna, zwykle dziedziczona. Rzadziej występuje spontanicznie (w wyniku spontanicznej zmiany genu = spontanicznej mutacji).
U chorych na hemofilię informacja genetyczna wymagana do wytworzenia funkcjonalnego czynnika krzepnięcia jest nieprawidłowa: w hemofilii A jest to czynnik krzepnięcia VIII, w hemofilii B czynnik IX. Skutkiem złego planu budowy jest to, że odpowiedni czynnik krzepnięcia nie może być wytworzony w wystarczająco funkcjonalnej ilości. To zakłóca krzepnięcie krwi: rany nie zamykają się tak szybko, więc krwawienie trwa niezwykle długo. Niektórzy pacjenci mają również spontaniczne krwawienia (bez wyraźnej przyczyny).
Hemofilia A i B: dziedziczenie
Plany (geny) wszystkich części ciała znajdują się na chromosomach. W jądrze każdej komórki ciała znajduje się 46 chromosomów, w tym dwa chromosomy płci, które między innymi określają płeć. Kobiety mają dwa chromosomy płci X (XX): Jeden chromosom X został odziedziczony po matce i jeden po ojcu. Z drugiej strony mężczyźni mają chromosom Y odziedziczony po ojcu i chromosom X odziedziczony po matce (XY).
Geny czynników krzepnięcia znajdują się na chromosomie X. U kobiet, które mają dwa chromosomy X, jeśli jeden z nich zawiera wadliwy schemat czynnika krzepnięcia, zwykle można to zrekompensować innym chromosomem X. W związku z tym są w dużej mierze bezobjawowe przez całe życie.
Jeśli taka kobieta ma dziecko, przekazuje potomstwu jeden z dwóch chromosomów X. Prawdopodobieństwo, że jest to kopia z wadliwym projektem, wynosi 50 procent. Przekazując defekt genetyczny, matka staje się nosicielką (nosicielką) hemofilii. Jeśli jest syn, urodzi się hemofiliakiem. Z kolei córka zwykle staje się potencjalną nosicielką.
W rzadkich przypadkach odpowiedni współczynnik krzepnięcia nie jest wystarczająco uformowany nawet w przewodach żeńskich. Rany po urazach lub operacjach mogą wówczas krwawić przez długi czas. Jeszcze rzadziej dziewczynka dziedziczy wadliwy chromosom X od obojga rodziców. Jest to związane głównie z dziedziczną chorobą zespołu Turnera. Chore dziewczęta ukazują pełny obraz hemofilii.
Zespół von Willebranda-Juergensa
W zespole von Willebranda-Jürgensa instrukcje dotyczące czynnika von Willebranda (vWF) wskazują na mutację: czynnik krzepnięcia jest za mały lub wadliwy. Mutacja genu może wystąpić zarówno u mężczyzn, jak iu kobiet.
Hemofilia: badania i diagnoza
Jeśli ktoś ma częste spontaniczne krwawienia (takie jak krwawienia z nosa) lub bardzo łatwo powstają siniaki, może to wskazywać na hemofilię. To podejrzenie jest szczególnie oczywiste, jeśli rodzina zna już przypadki hemofilii. Osoby dotknięte chorobą powinny mieć wyjaśnioną przez lekarza zwiększoną skłonność do krwawień. Pierwszym punktem kontaktu, jeśli podejrzewasz hemofilię, jest Twój lekarz rodzinny:
Lekarz najpierw zapozna się z historią choroby pacjenta (wywiad) w rozmowie z pacjentem: ma szczegółowo opisane objawy, pyta o choroby leżące u ich podłoża i czy są znane przypadki hemofilii w rodzinie.
Testy laboratoryjne są szczególnie ważne dla wyjaśnienia ewentualnej hemofilii. Lekarz pobiera od pacjenta próbkę krwi w celu przebadania jej w laboratorium pod kątem różnych parametrów: W hemofilii tak zwany czas częściowej tromboplastyny po aktywacji jest dłuższy niż u osób zdrowych. Natomiast tak zwana wartość Quick (czas tromboplastyny, TPZ) i czas trombinowy w osoczu (PTZ) są na ogół normalne; są przedłużone tylko w ciężkiej hemofilii. Liczba płytek krwi (trombocytów) i tzw. czas krwawienia są również prawidłowe w hemofilii A i B. Z drugiej strony w zespole von Willebranda-Jürgensa wydłuża się czas krwawienia. Czas krwawienia to czas potrzebny do zatrzymania krwawienia.
Aby móc jednoznacznie określić hemofilię A lub B, należy przeanalizować aktywność czynników krzepnięcia (VIII, IX). Odpowiada za to specjalista hematolog lub specjalistyczny ośrodek medyczny (centrum hemofilii).
Hemofilia: test u noworodków i nienarodzonych dzieci
Jeśli w rodzinie rozwinęła się już hemofilia, koagulacja noworodków płci męskiej jest zwykle sprawdzana natychmiast po urodzeniu. W ten sposób hemofilię można wykryć na wczesnym etapie. Możesz również sprawdzić hemofilię w czasie ciąży.
Jeśli kobieta podejrzewa, że ma genetyczne predyspozycje do hemofilii, a zatem jest potencjalnym nosicielem, można to wyjaśnić testem genetycznym.
Hemofilia: przebieg choroby i rokowanie
Hemofilia nie została jeszcze wyleczona. Pacjenci przez całe życie muszą radzić sobie z brakiem czynników krzepnięcia. Jednak przy pomocy koncentratów czynników mogą zwykle prowadzić w dużej mierze normalne życie.
Nieleczona, umiarkowana i ciężka hemofilia często prowadzi do poważnych powikłań. Na przykład krwawienie do mięśni może spowodować uszkodzenie mięśni. Krwotok w stawach może prowadzić do choroby zwyrodnieniowej stawów i usztywnienia stawów. Takich powikłań można uniknąć, jeśli hemofilia zostanie wcześnie wykryta i konsekwentnie leczona.
Tagi.: pragnienie posiadania dzieci spać opieka nad osobami starszymi





.jpg)