Zespół Marfana
Mareike Müller jest niezależną pisarką w dziale medycznym i asystentką lekarza neurochirurgii w Düsseldorfie. Studiowała medycynę człowieka w Magdeburgu i zdobyła wiele praktycznych doświadczeń medycznych podczas pobytów za granicą na czterech różnych kontynentach.
Więcej o ekspertach Wszystkie treści są sprawdzane przez dziennikarzy medycznych.Zespół Marfana (MFS) to choroba genetyczna tkanki łącznej. Pacjenci mają różne objawy w różnym stopniu: długie palce i wąskie, długie kończyny czy uszkodzenie naczyń krwionośnych. Nie ma lekarstwa na zespół Marfana. Regularne kontrole mogą zapobiec komplikacjom. Przeczytaj wszystko o zespole Marfana tutaj!
Kody ICD dla tej choroby: Kody ICD to uznane na całym świecie kody diagnoz medycznych. Można je znaleźć np. w pismach lekarskich czy na zaświadczeniach o niezdolności do pracy. Q87
Zespół Marfana: opis
Zespół Marfana jest chorobą genetyczną, która jest przenoszona z rodziców na dziecko lub rozwija się samoistnie. Choroba, która rozwija się samoistnie, znana jest również jako choroba sporadyczna. Dotyczy to około 25 do 30 procent pacjentów z zespołem Marfana. Ogólnie rzecz biorąc, od jednej do pięciu na 10 000 osób w populacji cierpi na zespół Marfana. Nie ma różnicy między płciami.
Zespół Marfana: objawy
Objawy zespołu Marfana są bardzo różne i różnie wyrażone u poszczególnych pacjentów. Nawet w tej samej rodzinie objawy zespołu Marfana mogą się znacznie różnić wśród chorych członków rodziny. Choroba wpływa na różne układy narządów. Najczęstsze są zmiany
- Układu sercowo-naczyniowego
- szkielet
- oko
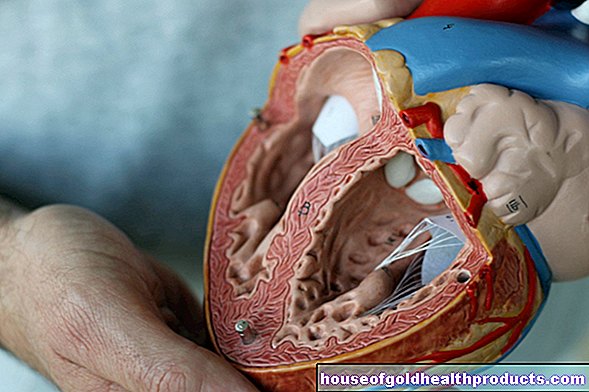
Zespół Marfana: Układ sercowo-naczyniowy
Pacjenci z zespołem Marfana są narażeni na zwiększone ryzyko nagłej śmierci. Powodem tego jest często występujące rozerwanie ściany tętnicy głównej (rozwarstwienie aorty). W wyniku powstania szczeliny w ścianie aorty krew nie jest już transportowana do mniejszych naczyń krwionośnych, ale raczej wnika do szczelin. Ryzyko rozwarstwienia aorty jest zwiększone u pacjentów z zespołem Marfana, ponieważ ich aorta, która ma osłabione ściany, stopniowo się poszerza (postępujące poszerzenie aorty).
Ponadto pacjenci często cierpią na uszkodzenia zastawek serca, takie jak niedomykalność zastawki aortalnej i mitralnej. Mogą one prowadzić do zaburzeń rytmu serca. Ponadto są narażeni na zapalenie serca (zapalenie wsierdzia) i niewydolność serca.
Zespół Marfana: szkielet
Zmiany szkieletowe są często pierwszą oznaką zespołu Marfana. Pacjenci wyróżniają się wysokim wzrostem i bardzo wąskimi, długimi kończynami. Palcowanie pająka (arachnodaktylia) jest dobrze znanym objawem. Palce pająka są nazywane takimi, ponieważ są niezwykle długie i wąskie.
Ponadto wielu pacjentów ma deformacje klatki piersiowej, takie jak pierś z kurczaka lub lejka. W miarę dalszych zmian kostnych często cierpią na skoliozę, zgięcie i skręcenie kręgosłupa. Ponadto niektórzy pacjenci mają słabo rozwinięte kości twarzy, takie jak kości policzkowe lub górna szczęka.
Całość tych zmian szkieletowych jest również znana jako habitus marfanoidalny.
Zespół Marfana: oko
Zmiany w oku spowodowane zespołem Marfana dotyczą głównie soczewki. Często jest przesunięty (ektopia soczewki). Grozi to pacjentowi ślepotą. Innym czynnikiem ryzyka ślepoty jest krótkowzroczność. Jest to spowodowane zbyt długą gałką oczną. Ta zmiana może również prowadzić do odwarstwienia siatkówki.
Zespół Marfana: objawy dotyczące innych narządów
Oprócz wspomnianych układów narządów, zespół Marfana może również uszkadzać inne struktury. Obejmuje to między innymi płuca. Osoby dotknięte chorobą mają zwiększone ryzyko rozwoju odmy opłucnowej. Lekarze rozumieją, że oznacza to oderwanie opłucnej płuc od opłucnej i wnikanie powietrza do tej szczeliny. Ten stan może zagrażać życiu, ponieważ płuca zapadają się w dotkniętym obszarze.
Rozstępy są często widoczne na skórze pacjentów z zespołem Marfana jako oznaka słabej tkanki łącznej.
W ciągu życia może rozwinąć się tzw. duraktazja. Jest to przedłużenie opon mózgowo-rdzeniowych, zwykle na poziomie odcinka lędźwiowego kręgosłupa. Często przebiega bezobjawowo. W niektórych przypadkach może powodować ból, gdy worek oponowy naciska na wychodzące nerwy rdzeniowe.
Zespół Marfana: przyczyny i czynniki ryzyka
Zespół Marfana jest autosomalną dominującą chorobą dziedziczną. Oznacza to, że zachodzi zmiana (mutacja) w genie w naszej strukturze genetycznej, która wywołuje chorobę. Autosomalna dominacja opisuje, że ta informacja genetyczna jest zlokalizowana w niespecyficznym dla płci kompleksie genów (autosomalnym) i zawsze pojawia się (dominująca).
Kiedy pacjent z zespołem Marfana ma dziecko, może odziedziczyć chory lub zdrowy gen. Ponieważ każda osoba ma podwójny zestaw genetyczny. Oznacza to, że prawdopodobieństwo transmisji wynosi 50 procent. Dziecko pacjenta z zespołem Marfana ma 50 procent szans na zachorowanie.
Zespół Marfana: uszkodzona tkanka łączna
Mutacja powodująca zespół Marfana znajduje się na długim ramieniu chromosomu 15 (15q21). Wpływa na tzw. gen FBN1. Gen ten jest odpowiedzialny za tworzenie białka tkanki łącznej, fibryliny-1. Fibrylina-1 jest ważna dla stabilności tkanki łącznej. Jeśli jego powstawanie jest ograniczone przez mutację, tkanka łączna traci stabilność.
Zespół Marfana: różne formy
Nasilenie zespołu Marfana jest różne. Lekarze mówią wtedy o zmiennej ekspresji. Oznacza to, że objawy pacjentów różnią się również w rodzinie. Mimo tej samej mutacji pacjent prawie nie może mieć żadnych objawów, podczas gdy rodzeństwo pokazuje pełny obraz zespołu Marfana.
Zespół Marfana: dochodzenia i diagnoza
Diagnozę zespołu Marfana często stawia pediatra. Ogólnie rzecz biorąc, różni specjaliści odgrywają rolę w diagnostyce, leczeniu i doradztwie. Oprócz pediatry obejmuje to genetyków ludzkich, kardiologów, ortopedów i okulistów. Przed ostatecznym postawieniem diagnozy lekarz najpierw zapyta Cię szczegółowo o Twoją historię medyczną (wywiad). Zada Ci m.in. następujące możliwe pytania:
- Czy członek rodziny ma zespół Marfana?
- Czy od czasu do czasu czujesz przyspieszone bicie serca?
- Czy zawsze byłeś wyższy od innych, kiedy byłeś dzieckiem?
- Czy jesteś krótkowzroczny?
Badanie fizykalne zespołu Marfana
Twój lekarz przeprowadzi wtedy badanie fizykalne. Czyniąc to, najpierw przygląda się bliżej szkieletowi. Zwraca uwagę na długość poszczególnych kości, kształt klatki piersiowej i kształt twarzy. Potem słucha serca i płuc. Nad główną tętnicą można zauważyć zaburzenia rytmu serca lub szumy przepływu.
W celu postawienia diagnozy zespołu Marfana opracowano tzw. kryteria Gent. Wymienia różne objawy choroby w różnych postaciach. Po spełnieniu określonej liczby kryteriów można postawić diagnozę.
Możliwy jest również test genetyczny na zespół Marfana. Analizowany jest skład genetyczny i poszukuje się mutacji odpowiedzialnej za chorobę. Jeśli w rodzinie występują przypadki zespołu Marfana, odpowiednią diagnozę można postawić przed porodem.
Zespół Marfana: Podobne obrazy kliniczne
Inne choroby genetyczne, które mogą prowadzić do podobnych objawów, należy odróżnić od zespołu Marfana. Należą do nich m.in.
- Zespół Ehlersa-Danlosa
- Zespół Loeysa-Dietza
- Zespół Sphrintzena-Goldberga
- zespół MASS
Zespół Marfana: leczenie
Ponieważ zespół Marfana jest chorobą genetyczną, sama przyczyna, mutacja, nie może być leczona. Celem terapii jest regularne badanie pacjenta przez różnych specjalistów w celu zapobiegania powikłaniom. Co najważniejsze, monitorowanie serca jest ważne, aby zapobiec nagłej śmierci z powodu rozwarstwienia aorty. Można to osiągnąć poprzez przeciwdziałanie rozszerzeniu aorty poprzez podawanie beta-blokerów i ograniczenie aktywności fizycznej. Rozszerzenie aorty można monitorować za pomocą corocznych badań USG, a korzeń aorty można skorygować odpowiednio wcześnie przed rozwarstwieniem.
Dalsze operacje, które mogą być konieczne w przypadku zespołu Marfana
- Korekcja skoliozy
- Korekcja klatki piersiowej
- Usuwanie soczewki
Zespół Marfana: przebieg choroby i rokowanie
Prawdopodobieństwo przeniesienia mutacji z jednego rodzica na dziecko wynosi 50 procent. Pary mające partnera z zespołem Marfana, które planują mieć dzieci, powinny zasięgnąć porady genetyka.
Obecnie oczekiwana długość i jakość życia pacjentów z zespołem Marfana są prawie nieograniczone. Chociaż w przeszłości średnia długość życia była wyraźnie ograniczona, w ciągu ostatnich 30 lat wzrosła o 30 lat. Jednak pacjenci nadal są narażeni na zwiększone ryzyko rozwarstwienia aorty, co może skutkować nagłą śmiercią. Rozwarstwienie aorty najczęściej obserwuje się w wieku około 30 lat. Regularne kontrole przez specjalistów prowadzących leczenie mogą zmniejszyć ryzyko rozwarstwienia aorty w zespole Marfana.
Tagi.: narkotyki alkoholowe włosy maluch












.jpg)