Zespół Angelmana
Lisa Vogel studiowała dziennikarstwo wydziałowe ze szczególnym uwzględnieniem medycyny i nauk biologicznych na Uniwersytecie Ansbach i pogłębiła swoją wiedzę dziennikarską, uzyskując tytuł magistra informacji i komunikacji multimedialnej. Następnie odbył się staż w redakcji Od września 2020 pisze jako niezależna dziennikarka dla
Więcej postów Lisy Vogel Wszystkie treści są sprawdzane przez dziennikarzy medycznych.Zespół Angelmana (Syndrom Szczęśliwego Lalka) jest rzadką chorobą genetyczną. Przejawia się m.in. ograniczeniami psychicznymi i fizycznymi, zaburzeniami rozwojowymi (zwłaszcza językowymi) oraz nadpobudliwością. Uderzający jest wygląd lalki i radosny wyraz twarzy osób dotkniętych chorobą. Dowiedz się więcej o rzadkiej chorobie!
Kody ICD dla tej choroby: Kody ICD to uznane na całym świecie kody diagnoz medycznych. Można je znaleźć np. w pismach lekarskich czy na zaświadczeniach o niezdolności do pracy. Q93

Krótki przegląd
- Co to jest zespół Angelmana? Rzadka choroba genetyczna objawiająca się ograniczeniami psychicznymi i fizycznymi w rozwoju dziecka
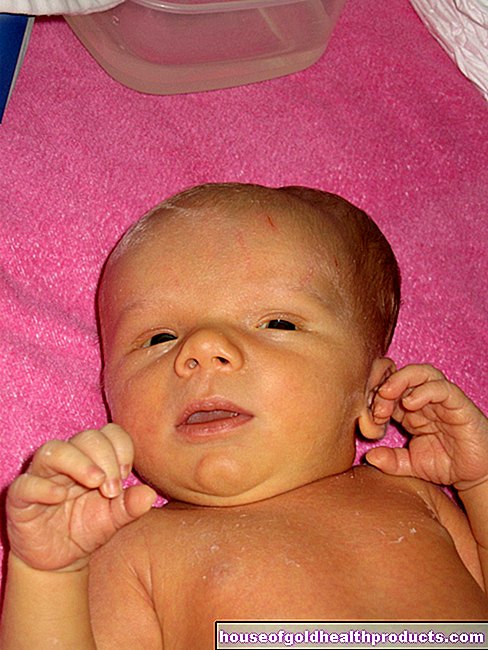
- Objawy: rysy twarzy przypominające lalkę, zaburzenia rozwoju, zaburzenia koordynacji, brak lub prawie żaden rozwój języka, obniżona inteligencja, drgawki, bezpodstawny śmiech, napady śmiechu, nadmierne ślinienie się, radosne machanie rękami
- Przyczyny: defekt genetyczny na chromosomie 15
- Diagnoza: m.in. rozmowa, badanie fizykalne, badania genetyczne
- Terapia: Brak dostępnej terapii przyczynowej; Wspomagające, np. fizjoterapia, logopedia, terapia zajęciowa; ewentualnie leki łagodzące objawy (np. w przypadku drgawek)
- Prognoza: normalna długość życia; nie ma możliwości samodzielnego życia
Zespół Angelmana: definicja
Zespół Angelmana (AS) jest spowodowany defektem genetycznym na chromosomie 15. Wada ta zakłóca rozwój fizyczny i umysłowy osób dotkniętych chorobą. Zaburzenia rozwoju mowy, niepewność ruchowa i szczęśliwa twarz to najbardziej zauważalne objawy zespołu Angelmana.
Nazwa „Syndrom Angelmana" pochodzi od odkrywcy choroby, angielskiego pediatry Harry'ego Angelmana. W 1965 roku porównał obrazy kliniczne trojga dzieci o rysach twarzy przypominających lalki. Dzieci dużo się śmiały i wykonywały szarpane ruchy - jak marionetki Doprowadziło to do angielskiej nazwy „ Happy Puppet Syndrome ”(szczęśliwa marionetka).
Zespół Angelmana występuje u obu płci. Ryzyko choroby wynosi około 1: 20 000. To sprawia, że zespół jest rzadką chorobą.
Zespół Angelmana: objawy
Po urodzeniu dzieci z zespołem Angelmana są normalne. Zaburzenia rozwoju motorycznego i poznawczego stają się coraz bardziej zauważalne w okresie niemowlęcym i wczesnym dzieciństwie. Cechy zaburzenia genetycznego to:
- opóźniony rozwój motoryczny
- zaburzona koordynacja
- często brak lub prawie żaden rozwój języka
- obniżona inteligencja
- nadpobudliwe, pobudliwe zachowanie
- bezpodstawny śmiech
- Napady śmiechu
- radosne gesty (np. machanie rękami)
- nadmierny ślinotok
- częste wystawianie języka
Niektóre dzieci z zespołem Angelmana mają również:
- Małogłowie (nienormalnie mała głowa) - nie przy urodzeniu, ale w miarę rozwoju
- Napady padaczkowe
- Zmiany w aktywności elektrycznej mózgu
- bardzo jasna skóra i oczy z powodu zmniejszonej pigmentacji (hipopigmentacja)
- Zez (zez)
Zespół Angelmana: przyczyny
Przyczyną zespołu Angelmana jest defekt genetyczny na chromosomie 15: funkcja genu UBE3A jest upośledzona u osób dotkniętych chorobą. Ten gen normalnie wytwarza enzym, który bierze udział w rozkładaniu uszkodzonych lub zbędnych białek w komórkach. W ten sposób pomaga komórce normalnie pracować.
Gen UBE3A znajduje się w regionie chromosomu 15q11q13. Tam geny podlegają tak zwanemu „wdrukowaniu genomowemu”. Oznacza to, że są one aktywne tylko na jednym z chromosomów rodzicielskich (w komórkach naszego organizmu znajdują się dwie kopie każdego chromosomu – jedna od matki i jedna od ojca). Reguluje to proces chemiczny - metylacja: przyłączone w określonych punktach grupy metylowe mogą włączać lub wyłączać gen.
Gen jest aktywny na obu chromosomach w wielu komórkach ciała, ale nie w komórkach nerwowych mózgu: u wielu tam ludzi gen UBE3A na 15 chromosomie ojcowskim jest wyłączany przez imprinting. W rezultacie UBE3A jest aktywny tylko na matczynym chromosomie 15 w mózgu. Oznacza to również: Jeśli matczyna kopia genu wykazuje błąd, nie można tego zrekompensować nieużywaną ojcską kopią genu. I to właśnie ta kombinacja występuje w zespole Angelmana: segment genu ojcowskiego jest wyłączony, segment genu matczynego jest uszkodzony.
Podstawowy błąd genetyczny może być różnego rodzaju:
- Usunięcie: Około 75 procent wszystkich osób z zespołem Angelmana nie ma odpowiedniego regionu 15q11q13 z genem UBE3A na chromosomie 15 matki. Ponieważ odpowiedni region na chromosomie 15 ojcowskim jest „wyłączany” przez imprinting, organizm może użyć enzymu, którego Plan Konstrukcji jest zapisany w Gen UBE3A, nie twórz.
- Mutacja w genie UBE3A: Samorzutnie zachodząca zmiana w genie powoduje utratę informacji w nim zawartych. Dotyczy to od pięciu do dziesięciu procent osób z zespołem Angelmana. W mniej więcej co piątym przypadku występuje mutacja rodzinna: tutaj matka nosi już zmianę genetyczną na chromosomie ojca.
- Dwa ojcowskie chromosomy 15: Osoba dotknięta chorobą odziedziczyła oba chromosomy 15 od ojca, żaden od matki (medycznie określana jako „ojcowska jednorodzicielska disomia 15”). Nie ma więc aktywnego genu UBE3A. Dotyczy to około jednego do dwóch procent wszystkich pacjentów z zespołem Angelmana.
- Błąd imprintingu: Gen UBE3A na matczynym chromosomie 15 - podobnie jak ten na ojcowskim chromosomie 15 - jest wyłączany przez imprinting. Ponadto może brakować pewnej części chromosomu (skreślenie). Błąd odcisku występuje w jednym do czterech procent przypadków zespołu Angelmana.
W pozostałych dziesięciu do 15% przypadków przyczyna zespołu Angelmana jest nieznana. Przy okazji: jeśli gen matczyny jest wyłączony, a gen ojcowski jest wadliwy, dzieci cierpią na tak zwany zespół Pradera-Williego.
Czy zespół Angelmana jest dziedziczny?
Ogólnie rzecz biorąc, ryzyko nawrotu w zespole Angelmana jest niskie. Oznacza to ryzyko, że rodzice chorego dziecka będą mieli inne dzieci, które również mają ten zespół. Jednak w indywidualnych przypadkach ryzyko to w dużej mierze zależy od wady genetycznej leżącej u podstaw zespołu Angelmana.
Na przykład w zespole Angelmana z powodu dwóch chromosomów ojcowskich 15 (jednorodzicielska disomia ojcowska 15) jest to mniej niż jeden procent. Z drugiej strony zespół Angelmana z powodu błędu imprintingu z utratą pewnego segmentu genu (delecja IC) może wystąpić w połowie wszystkich przypadków u rodzeństwa.
To zwiększone ryzyko istnieje również w przypadku mutacji UBE3A - pod warunkiem, że matka jest już nosicielką wady genetycznej (w około 20 procentach wszystkich przypadków mutacji). W takich przypadkach matka odziedziczyła mutację po ojcu. UBE3A ulega zatem zmianie w ojcowskim chromosomie matki. Jeśli to jest wyłączone, matka nie zauważy mutacji. Może jednak przekazać chromosom swoim dzieciom, gdzie – wtedy jako chromosom matczyny – może wywołać zespół Angelmana.
Teoretycznie pacjenci z zespołem Angelmana mogą się rozmnażać. W zależności od tego, kiedy wystąpiły przyczynowe zmiany chromosomowe (np. już podczas rozwoju komórek zarodkowych lub bezpośrednio po zapłodnieniu), ryzyko jest czasami bardzo wysokie (do 100 procent), że osoby dotknięte chorobą przeniosą chorobę. Brakuje jednak wiarygodnych danych na ten temat. We wrześniu 1999 r. zdarzył się odosobniony przypadek: matka, która miała zespół Angelmana, przekazała chorobę tutaj.
Zespół Angelmana: diagnoza
Jeśli zauważysz u dziecka opisane powyżej objawy, pierwszym punktem kontaktu jest pediatra. Może on dokładniej zawęzić możliwe przyczyny i w razie potrzeby skierować Ciebie i Twoje dziecko do specjalisty.
anamnese
Pierwszym krokiem w diagnozie jest dokładna historia choroby. Lekarz zada Ci różne pytania dotyczące Twojego dziecka, takie jak:
- Jakie zmiany zauważyłeś u swojego dziecka?
- Czy Twoje dziecko ma jakieś dolegliwości fizyczne?
- Czy Twoje dziecko może siedzieć?
- Czy Twoje dziecko sięga po przedmioty?
- Czy Twoje dziecko mówi?
- Czy Twoje dziecko często jest wyraźnie radosne lub podekscytowane?
- Czy Twoje dziecko śmieje się w nieodpowiednich sytuacjach, na przykład gdy odczuwa ból?
Badanie lekarskie
Po tym następuje badanie fizykalne. Pediatra sprawdza, w jakim stopniu dziecko regularnie rozwija zdolności motoryczne i umysłowe. Służą do tego proste ćwiczenia: np. dziecko powinno skoncentrować się na zabawkach lub wybiórczo sięgać po klocek. Lekarz zwraca również uwagę na mimikę dziecka. Częsty śmiech, rysy przypominające lalkę i ślinotok to oznaki syndromu Angelmana.
Jeśli po badaniu fizykalnym istnieje podejrzenie rzadkiej choroby, lekarz skieruje Cię do neurologa i genetyka ludzkiego.
Testy genetyczne
Testy genetyczne są ważną częścią diagnozowania zespołu Angelmana. Lekarzowi potrzebna jest niewielka próbka komórek dziecka, którą może pozyskać z błony śluzowej jamy ustnej, np. pobierając próbkę krwi lub wymaz. Materiał genetyczny (DNA) tych komórek lub odpowiedni region chromosomu 15q11q13 jest następnie badany bardziej szczegółowo w laboratorium.
W pierwszym kroku lekarze zwracają uwagę na wzór metylacji segmentu chromosomu (analiza/test metylacji). Dalsze badania na tych samych próbkach (analiza delecji, analiza mutacji) pomagają dokładniej określić przyczynę zespołu Angelmana. W tym celu konieczne może być również zbadanie składu genetycznego rodziców. W ten sposób lekarze mogą ustalić, czy istnieje już tam defekt genetyczny.
Dalsze dochodzenia
Często przydatne są dalsze badania. Na przykład EEG można wykorzystać do wykrywania zmian w aktywności elektrycznej mózgu, co często ma miejsce w zespole Angelmana. Wskazane mogą być również badania okulistyczne.
Zespół Angelmana: Terapia
Zespół Angelmana jest nieuleczalny – genetycznej przyczyny choroby nie da się usunąć. Jednak wczesna interwencja może mieć pozytywny wpływ na rozwój motoryczny i umysłowy osób dotkniętych chorobą. Pomocna jest na przykład regularna fizjoterapia. Może poprawiać zdolności motoryczne dzieci, łagodzić ograniczoną mobilność i zapobiegać chorobom wtórnym, takim jak deformacje kręgosłupa. Dzieci z zespołem Angelmana korzystają również z innych metod terapeutycznych, takich jak terapia zajęciowa i logopedia.
Ponadto niektóre objawy i stany związane z zespołem Angelmana mogą wymagać ukierunkowanego leczenia. Na przykład leki przeciwdrgawkowe (leki przeciwpadaczkowe) pomagają w napadach padaczkowych, a środki uspokajające (uspokajające) pomagają w ciężkich zaburzeniach snu.
Na stronie internetowej Verein Angelman e.V. znajdziesz wiele informacji na temat zespołu Angelmana, relacje z doświadczeń i wydarzeń dla poszkodowanych, a także osoby kontaktowe dla poszkodowanych we wszystkich regionach Niemiec.
Zespół Angelmana: przebieg choroby i rokowanie
Pierwszy rok życia
Dzieci z zespołem Angelmana częściej mają problemy z karmieniem piersią, ssaniem i połykaniem. Często wystawiają języki lub bardzo się ślinią. Ponadto dzieci z zespołem Angelmana często plują (co często mylone jest z nietolerancją pokarmową lub chorobą refluksową). Częste wymioty mogą prowadzić do niebezpiecznej utraty wagi.
W wieku od trzech do sześciu miesięcy dzieci z zespołem Angelmana często zaczynają się uśmiechać. Często chichoczą i bulgoczą, a podczas tych wybuchów radości często wystawiają języki.
Opóźniony rozwój motoryczny jest zwykle zauważalny w wieku od 6 do 12 miesięcy: dzieci nie raczkują ani nie siadają. Ruchy górnej części ciała są często chwiejne. To z kolei utrudnia siedzenie.
Część osób dotkniętych chorobą ma już napady padaczkowe w wieku 12 miesięcy.
Od roku do trzech lat
W ciągu pierwszych trzech lat życia zaburzenia rozwojowe w zespole Angelmana stają się bardzo widoczne. Dzieci cierpią bardziej na łagodne napady padaczkowe. Niektórzy z nich są nadpobudliwi, nadmiernie podekscytowani i zawsze w ruchu. Wiele osób ma tendencję do ciągłego wkładania rąk lub zabawek do ust lub częstego wystawiania języka i ślinienia się. Jeśli dzieci są szczególnie podekscytowane, często nadmiernie się śmieją i machają z wyciągniętymi ramionami.
Zaburzony rozwój języka staje się widoczny po raz pierwszy w tym wieku. Dzieci „gaworzą” do siebie lub krzyczą i piszczą, jednak potrafią wymówić tylko kilka lub wcale nie zrozumiałych słów i zwykle używają ich bez kontekstu.Ale rozumieją język i są też interakcje społeczne z innymi ludźmi.
Dojrzewanie i dorosłość
U dzieci z zespołem Angelmana dojrzewanie jest często opóźnione o trzy do pięciu lat. Jednak dojrzałość płciowa rozwija się wtedy normalnie. Nadal nie ma rozwoju języka, jeśli często obecne jest rozumienie języka. Napady padaczkowe w wieku dorosłym można zwykle dobrze leczyć za pomocą leków.
Osoby z zespołem Angelmana mają normalną długość życia. Niezależne życie nie jest możliwe ze względu na ograniczenia psychiczne.
Tagi.: Dziecko Dziecko pierwsza pomoc alkohol